- 全站
- 商品
- 资讯
河南时事通医疗器械咨询有限公司

打造医疗器械创新产品孵化加速器
致力于医疗器械产品有效性及安全性试验研究工作
提供从质量体系建立到产品注册的全流程技术服务
药物临床试验流程图(药物临床试验全流程梳理)
临床研究在整个药物研发过程起着决定性的作用,而临床试验作为重要的表现形式,一方面可以对新药审评和注册提供法规要求的申报资料,为企业研发新药、明确市场战略决策提供依据,另一方面可以评价新药潜在的临床应用价值、确定新药的更佳使用方法,为广大患者带来福音。因此,本文针对临床试验全流程进行整合阐述。
临床试验全流程
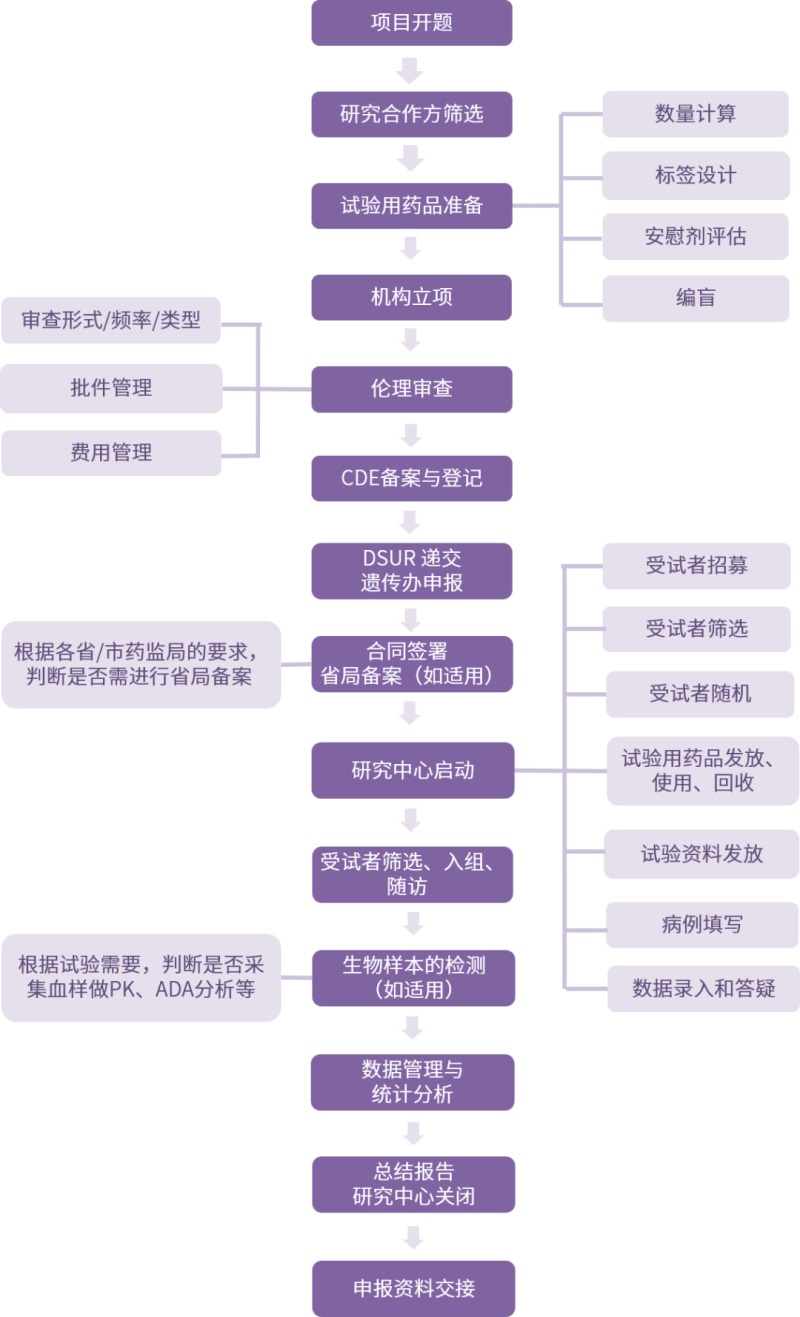
临床试验复杂而漫长,涉及环节众多,细节繁琐,在试验过程中对各环节内容、各中心制度流程及相应的法律法规的熟悉程度,决定了临床试验进程的快慢,以下总结了有关研究合作方筛选、机构立项、伦理审查、研究中心关闭等环节的具体内容及相关要求。
01研究合作方筛选
研究合作方筛选主要包括研究中心筛选和供应商筛选:
(1)研究中心筛选
由于临床研究涉及人体的临床医学研究,对于研究中心的选择显得尤为重要,选择的条件主要包括:研究中心是否具有入组受试者的能力及能承担的病人数量、医疗设施是否符合要求、是否具有足够的设备和相关的研究人员、是否具有临床研究经验;研究者是否具备专业领域及行政管理的资质等。
(2)供应商筛选
供应商筛选主要针对数据管理单位、统计分析单位、生物样本检测单位、SMO、招募、物流等进行选择。
02机构立项
在机构申请立项时,需通过各种渠道主动获取立项流程和相关要求,有针对性的进行相关文件的准备,明确相关的要求,包括资料份数、盖章、装订要求、各阶段时限等,以便于能够高效的与研究者进行沟通,顺利推进临床试验进程。
03伦理审查
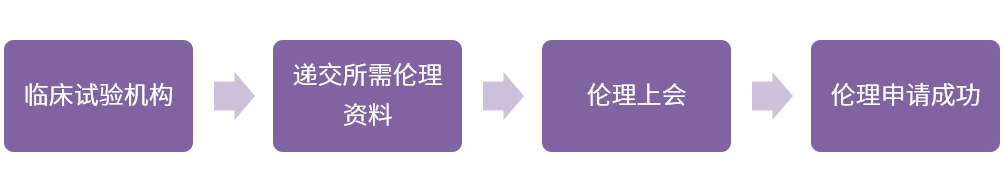
(1)咨询伦理相关内容
◆ 递交资料的清单、数量及特殊要求,例如装订形式、打印要求、是否盖章等;
◆ 接收资料的最后期限、伦理上会是否需要提供PPT、PPT讲解人、伦理会监查员是否可以参加;
◆ 伦理会召开的频率及最近一次召开的日期、获得伦理批件的时间;
◆ 伦理审评费、付款方式、医院帐户、获取发票的流程等。
(2)伦理资料递交的准备
◆ 准备伦理递交信,递交文件列表处要正确列出版本号、版本日期;
◆ 为PI准备伦理上会PPT;
◆ 所有递交给伦理委员会审批的文件必须是最终版的,应有主要研究者签字;
◆ 伦理委员会审批后的文件,如有任何更改,都必须通知伦理委员会备案。
(3)合同
◆ 在筛选中心时和各机构确定合同模板、签订流程及合同签订顺序;
◆ 了解机构收取管理费用比例;
◆ 筛选中心开始到各分中心伦理通过或伦理备案完成后;
◆ 合同审核工作,更好与伦理递交、审核同时进行;
◆ 分中心伦理通过后签合同;
◆ 签定合同后付第一笔款。
04CDE登记
CDE登记时间:
◆ 首例受试者签署知情同意书前;
◆ 信息更新后20个工作日内(如首例受试者签署知情同意书,方案、伦理等信息变更等);
◆ 因安全性原因主动暂停或者终止临床试验后10个工作日内;
◆ 项目完成后12个月内。
05研究中心启动
研究中心启动主要分为启动前准备、启动实施、启动跟进三部分。
(1)启动前准备
该阶段需进行研究机构硬件设备确认,试验用药品、物资确认,建立研究文档,发送启动访视确认信、申请启动费用及准备培训资料等。
(2)启动实施
该阶段需针对临床试验方案、试验用药品的管理(存储、发放、配置、使用、回收、销毁、异常(如超温)情况处理等)、合并用药的管理(允许使用和禁止使用的药品)、CRF填写或eCRF录入指南等与相关人员进行培训,同时进行研究人员资质、培训资料等收集。
(3)启动跟进
跟踪启动会过程中的未解决问题,撰写并发送启动访视报告及随访信。
06数据管理与统计分析
(1)数据管理
首先需撰写数据管理计划(第一稿在首例受试者签署知情同事书前定稿),由临床数据管理员进行数据的审核及清理,后召开数据审核会(如是盲法试验,还需进行盲态审核),形成数据审核报告,锁定数据库,最后撰写数据管理报告。
(2)统计分析
在数据库锁定之前,撰写统计分析计划并定稿,在数据库锁定后,进行第一次揭盲,撰写统计分析报告。
07研究中心关闭
确定试验资料将存放的条件和归档程序、研究者文件夹中的资料完整性 ,清点中心的入组受试者数量、清点试验药品、相关文件,将临床研究文件归档,并组织安排将药物返还给申办者等。
来源:新领先药讯
关
注
微
信
二
维
码

手
机
网
站
二
维
码
版权所有:河南时事通医疗器械咨询有限公司 豫ICP备2023006270号-1 营业执照
医疗器械经营备案 医疗器械生产证代办 二三类医疗器械注册办理 郑州医疗器械许可证